I just came back from the Keystone conference on the microbiome and metabolism in Banff, Canada, where Eran Elinav of the Weizmann Institute of Science presented research on the relationship between high blood sugar and the microbiome, published this week in the journal Science. (Click on the image below for the article.)
 The very quick summary of the paper is this: high blood sugar causes a leaky gut barrier and changes the gut microbiota. Those changes increase the chance of a gut infection.
The very quick summary of the paper is this: high blood sugar causes a leaky gut barrier and changes the gut microbiota. Those changes increase the chance of a gut infection.
One take home point – that diabetes and the gut microbiota are inextricably linked – is an important message for clinicians.
The other main message of the paper – that high blood sugar is a cause of gut dysbiosis (an unhealthy gut microbial community) and gut barrier leakiness – is interesting, but it implies that a metabolic defect in our body (high blood sugar or hyperglycemia) is the root cause of altered microbiota and leaky gut. This idea can be summarized as “you are the problem, not your gut microbes”.
The idea that high blood sugar is the underlying problem that causes leaky gut and infection contradicts work done by Patrice Cani and Nathalie Delzenne and others. Cani et al proposed the idea of metabolic endotoxemia.
Cani and Delzenne’s idea was that the Western diet causes dysbiosis and barrier dysfunction. Movement of bacteria products (“endotoxin” also known as lipopolysaccharide or LPS) and bacteria through a leaky gut barrier is proposed to drive low grade inflammation that is the cause of glucose intolerance and elevated blood sugar in patients.
Metabolic endotoxemia – an idea that has been refined in recent years – is an explanation for obesity, inflammation, and elevated blood sugar. The direction of causation is the opposite of Elinav’s argument. In other words, the gut microbiota is the problem (not you).
The figure below illustrates the concept. Source: Cani and Delzenne 2009 The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des 15(13) 1546-1558
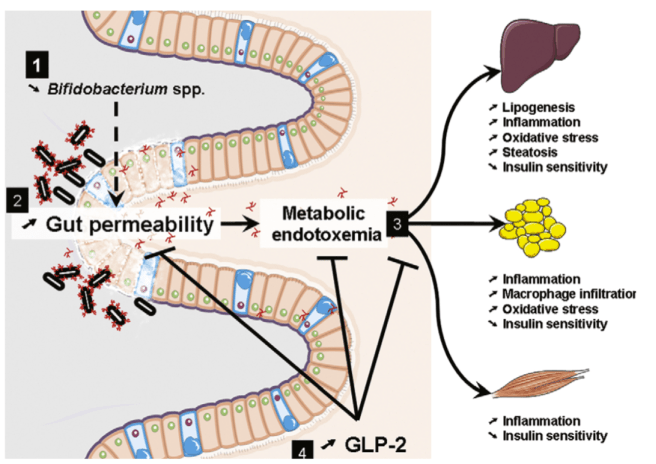
Which idea is correct? To me, the metabolic endotoxemia model fits what is known about diet and insulin resistance, and the hypothesis is supported by many studies involving the transfer of microbes to germ free mice. Known dietary associations with obesity support a role for metabolic endotoxemia in overweight and obesity.
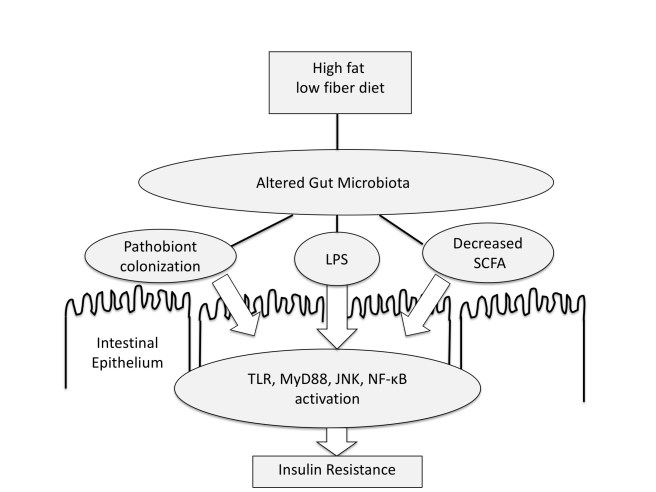
Why does this matter? If Elinav is right, and hyperglycemia is the main problem, then the way to prevent infection is to target is the defective host metabolism and make glucose levels normal e.g. with insulin. The problem is that intensive insulin treatment does not really work to improve outcomes in people. We covered how more insulin in the NICE SUGAR trial increased mortality in critically ill patients with infection.

On the other hand interventions that target the microbiome – not the host response – do show the potential for benefit. Probiotic treatment has a good track record of reducing enteric infection in humans. Probiotics also reduce blood sugar, but they do this by changing the composition of the gut microbiota and by reinforcing the gut barrier – consistent with Cani and Delzenne’s pioneering work.
Of course, antibiotics also target the gut microbiome and short term improvements in blood glucose can be accomplished by giving antibiotics to mice and to people.
It is possible of course that the core observations of both groups may be simultaneously correct. Diet and dysbiosis might cause barrier permeability and elevate blood sugar. And high blood sugar may alter the gut microbiota and gut barrier. Accurately predicting these events will be key to developing useful therapeutics.
How shall we accomplish that? This will require reductionist molecular biological approaches of the sort that Elinav’s laboratory is conducting, along with evolutionary approaches that uncover the selective pressures on microbes and mammals that shape our metabolism.
A variety of evolutionary hypotheses for the function of insulin resistance have been proposed. We have covered a variety of these on this blog, for example here, here, and here. For example, in Alcock and Lin 2015, we wrote
“We further propose that the modulation of metabolism by fats and microbiota may be adaptive in fueling the increased energy needs of immune cells activated by dysbiosis. By blocking glucose uptake, insulin resistance reduces energy utilization by tissues dependent on GLUT4 glucose uptake (predominantly skeletal muscle and fat) and diverts energy access to tissues not reliant on insulin-stimulated GLUT4. Phagocytes (for example, macrophages) and intestinal epithelial cells do not rely on GLUT4. As a result, glucose energy is expected to be preferentially delivered to activated innate immune cells in the gut during the insulin resistant state.”
The idea that insulin resistance and changed glucose metabolism might be adaptive in fueling immune cells during dysbiosis has yet to be explicitly tested. This hypothesis also does not explain all the findings of Elinav’s group. Another idea, developed by Ed Legrand and myself and others, suggests that the osmotic stress of high blood sugar increases the vulnerability of pathogens to other host defenses like fever and low pH, e.g. here. The merits of these hypotheses need more testing of course. More to come on this topic on this blog.
Categories: Uncategorized
Joe Alcock
Emergency Physician, Educator, Researcher, interested in the microbiome, evolution, and medicine

Fascinating stuff, Joe. My husband (Rob Coberly, whom you may remember from the VA) and I are very interested in mitochondrial dynamics and especially in how mitochondrial populations evolve in any given body according to selection pressures over the lifespan. Great summary article here : https://www.frontiersin.org/articles/10.3389/fnene.2013.00007/full. I think it may relate to what you are discussing here, since variations in available substrates for mitochondrial respiration could exert selection pressures both in the host body and in the biome. Glucose, fatty acids, amino acids, and ketone bodies can all serve as respiratory substrates but have different effects and byproducts, with consequent effects on host fitness.
I’m also interested in your diagram of insulin resistance above. I’m interested in your thoughts about how different the dynamic might be given input of a ketogenic diet high in green vegetables, i.e. relatively high in fat and protein but also very high in soluble and insoluble fibers and low in carbohydrates. This would most closely approximate the true Cretan diet, as opposed to the mishmash vaguely defined “Mediterranean” diet that is so often recommended. Evolution occurs within the human lifespan in the sense that our sub-populations evolve in response to selection pressures, and the line of research about how to exert positive selection pressures may be useful in medicine.
Thanks Heather for this thoughtful comment
Joe. Great summary. thank you and such an interesting area!
question: surely you can approach hyperglycemia by reducing carbohydrate? Why is insulin the only approach? Totally agreed there are a lot of issues with insulin treatment?