In 2014, Carlo Maley, Athena Aktipis, and I wrote an article on eating behavior and the gut microbiota that was published in Bioessays. We proposed that unhealthy food preferences, cravings and aversions may serve the evolutionary interests of microbes in our guts, sometimes in conflict with our own interests. We suggested that gut microbes potentially could hijack nervous system signaling by manufacturing neurotransmitters and appetite peptides. Indeed, gut microbes manufacture neuropeptides and hormone that are nearly or exactly the same as our own hormones and reward neurotransmitters. This gives microbes the means to manipulate us. We reasoned that genetic conflicts of interest around eating behavior provide the motive for microbes to hijack our eating behaviors. If true, what and how much we eat may be less an issue of willpower, and more the result of microbes in our bodies.
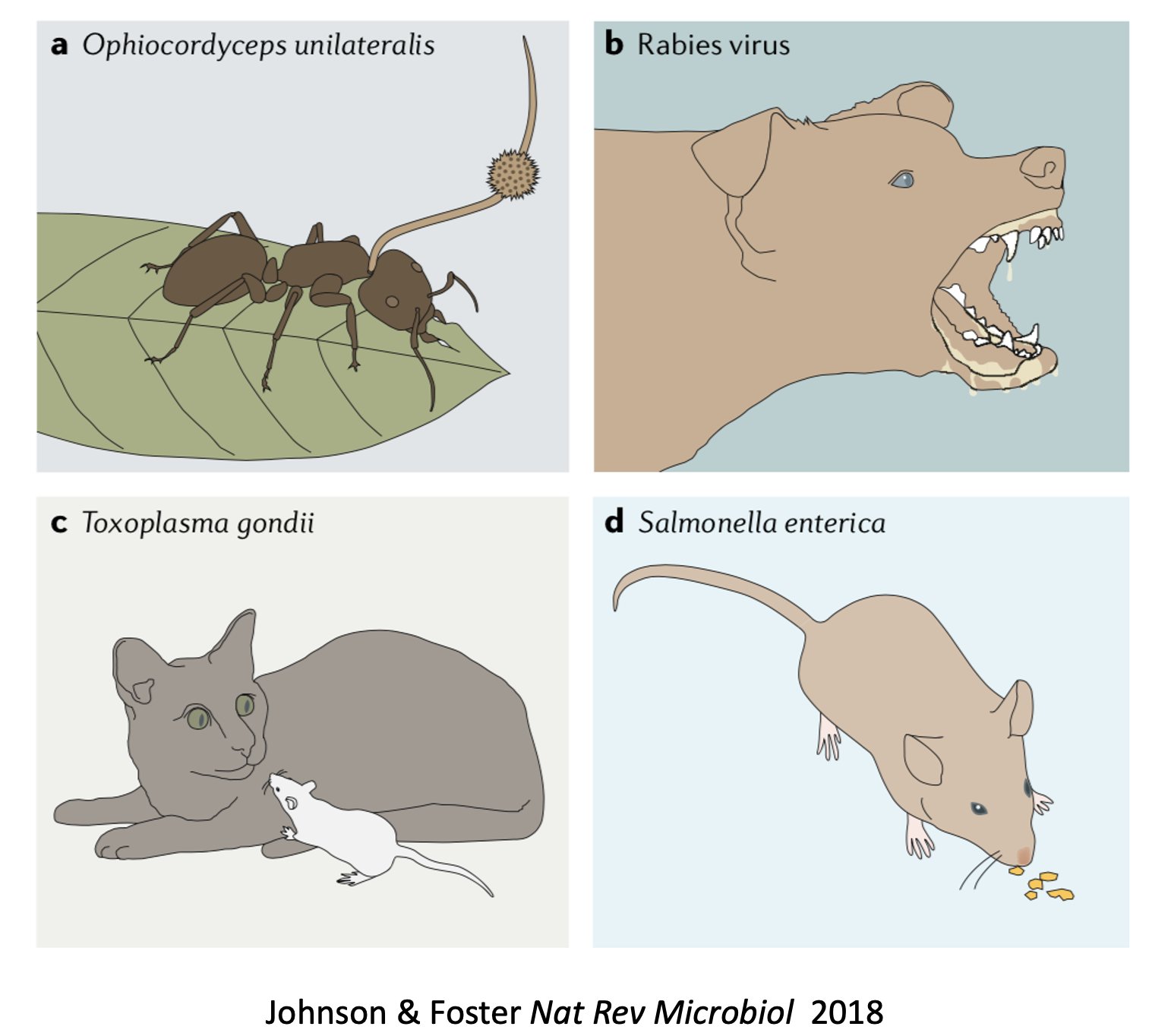
Examples of brain hijacking microbes
In biology, numerous examples exist of microbes manipulating the behavior of host organisms. Toxoplasmosis is the most commonly cited example. Mice with toxoplasmosis lose their fear of cats and cat urine. This is good for the Toxoplasmodium gondii parasite, and bad for the mouse which is more readily eaten by its feline predator. Might gut bacteria be capable of similar manipulation? We hypothesized that microbes influence eating behavior, in part by increasing the abundance of reward hormones when we consume the growth substrates that benefit those microbes. We also highlighted the possibility that microbes engage in a kind of blackmail, making us feel bad if we do not provide a constant supply of growth-limiting nutrients. If mood is controlled at some level by the microbes in your guts, how might that affect eating? Many studies have shown that gut bacteria induce anxiety and depression-like behavior in animals and humans. One possibility is that anxiety can result in stress eating that benefits the microbes that cause anxiety. On the other hand, several studies have shown that probiotics lessen anxiety and depression, and could be useful in preventing unhealthy eating. We further suggested that some modern trends we observe, around overeating, obesity and diabetes, may lie less in our genes or our brains, and more so in the genes of our gut microbes.
This work was inspired by evolutionary theorists, such as David Haig, who have highlighted puzzling features in a variety of diseases, that have at their core a genomic conflict of interest. One major area of conflict involves reproductive conflict causing common diseases in pregnancy. Another important example is cancer. For instance, mutations in somatic cell lines cause unrestrained replication that serves the short term fitness interests of the clonal cancer lineage that depart from the fitness interests of the rest of the body.
Similarly, we humans have different fitness interests than do our resident microbes, collectively known as the human microbiota. Aktipis, Maley and I highlighted conflicts of interest between microbe and human that arise from the fact that we do not share genes with resident microbes. Even though non-related organisms often cooperate, these shared allegiances are inherently fragile. When genetic differences exist, some conflict between unrelated organisms is unavoidable. In the case of the gut microbiota, what you eat has enormous effects on the which microbes benefit, can reproduce in numbers that permit a long-term residence in the guts, and allow them to outcompete competitors.
Food-dependent replication of microbes suggests that if any microbes had a capacity to affect human eating behavior, it would have an enormous fitness advantage. We would expect those sorts of manipulative features to evolve. We would also expect that humans would evolve ways to resist manipulation. Put differently, there might be a constant tug of war between ourselves and our microbes involving appetite hormones, activity in the gut, and in our brains.
Do microbes, in fact, control our food cravings in the way that we predicted in our 2014 paper? Research a decade ago showed that a preference for chocolate is linked with a specific microbiome and metabolite profile. This study showed an intriguing association, but did not prove that the microbes are responsible for preference for chocolate. The causal direction (if it exists) might plausibly also go in the other direction. Since that chocolate study was published, our group has explored whether the oral microbiota can linked with sweet consumption. Our results, yet unpublished, did not point to any breakthroughs. A more recent study in humans also looked for differences in taste perception linked with the oral microbiota. They did find a positive association. Additional work, reviewed here, found that the perception of food aromas is deeply tied to the presence and activity of oral microbes. Does this prove that oral microbes hijack taste receptors? No, but it remains a possibility.
In other species, microbes do appear to affect cravings. In rats, the microbiome transfered at birth from mother to offspring was recently shown to have an effect on the rat’s eating behavior. This effect of maternal microbiome transfer to offspring suggests that an animal’s development and later food preferences may be impacted by microbial or metabolite exposures very early in life. A recent study involving dietary “cravings” of fruit flies found that the microbiome can control whether an animal seeks out a specific amino acid missing from the diet. Boosting the numbers of certain microbes was able to override a fly’s “craving” for deficient nutrients. The microbiome of fruit flies also affects their sense of smell and whether they forage for microbes in addition to food. Fruit flies’ foraging preference for microbes is driven by the composition of the microbiome and by their early life microbial experience. One wonders if something similar happens in humans. Do we seek out microbe-rich foods, especially fermented foods that we were exposed to early in life? Such an idea is plausible, especially since fermented foods are widespread in human ecology.
How about other aspects of eating behavior, such as appetite and satiety? In one study, mice infected with Salmonella typhimurium ate more and pooped more, with the consequence of increasing disease transmission by increasing the amount of Salmonella in the infected animal’s poop. The experimenters reported this had no untoward effect on the infected mice, since eating more food seemed to increase their survival. Even so, this is an example of eating behavior being manipulated by a gut microbe that promotes the microbe’s fitness.
In another example of bacterial influence on eating behavior, E. coli and other bacteria produce a protein, ClpB, that is nearly identical to a hormone made by humans that regulates appetite, α-MSH. ClpB appears to cause an increase in satiety, or fullness, and a decrease in eating. Serguei Fetissov has argued that ClpB is a signal from bacteria that helpfully tells the host when to stop eating. In his view, this is an example of a mutualism, when both parties benefit from the communication between mammal and microbe. I disagree. I am skeptical that E. coli would benefit by cutting off its growth limiting nutrients. It is the host that benefits by stopping eating when a bacterial cue of excessive growth is detected. Why does the microbe produce the protein? ClpB is a heat shock protein that increases bacterial resistance to stress. In many bacteria, ClpB promotes virulence and makes the bacteria resistant to immune attack. As my collaborator Ed Legrand has pointed out, harmful microbes are more vulnerable to immune defenses when they are rapidly dividing (like E. coli in the rapid growth phase) than when they are quiescent. After the rapid growth phase (which is when ClpB is expressed) bacteria have the opportunity to shore up their defenses, which E. coli apparently does. Bacteria produce ClpB to prepare themselves for battle, not to tell us to stop eating. Under those circumstance, it should be no surprise that hosts pay close attention to ClpB and respond by slowing nutrient delivery and generating defensive pro-inflammatory cytokines. Again, this is not what we’d expect from a helpful and friendly chat between old friends.
It is notable that patients after weight loss surgery, such as Roux-en-Y surgery, show a marked increase in ClpB, in part because of bacterial overgrowth in the small intestine. Farin and colleagues found that after Roux-en-Y surgery, “the gene coding ClpB was found to be increased by 100 after LRYGB (P< .05, Wilcoxon signed-rank test; not shown), in parallel with the increase of E. coli that encodes it.” It is possible, therefore, that decreased satiety and the weight loss from Roux-en-Y happens because it induces a pathogenic state – small intestinal bacterial overgrowth. Conflict, not cooperative dialog, defines the relationship between mammal, microbe, and appetite regulating bacterial peptides such as ClpB. (ClpB also antagonizes appetite by increasing host peptide YY, or PYY).
Interestingly, bacterial ClpB is thought to elicit auto-antibodies against α-MSH. Autoantibody production against α-MSH, because of molecular mimicry with ClpB, has been proposed to be important in the pathophysiology of eating disorders. Elevated plasma levels of both ClpB and α-MSH auto-antibodies have been reported in eating disorder patients, although auto-antibodies to α-MSH are also detected in healthy individuals without eating disorders. That antibodies are made against ClpB is a clue that this microbial peptide is not benign for the host. Two potential reasons exist for antibody production. First, the effect may antagonize microbial interference with endogenous host signaling. Second, antibodies may diminish the function of this chaperone protein that permits bacteria to survive host defenses. These antibodies highlight conflict that has long existed during the coevolution of human and the microbiota.
Microbial metabolites are another important means by which gut microbiota influence eating. Fiber fermentation by gut bacteria produce short chain fatty acids (SCFA). The short chain fatty acids butyrate, propionate, acetate are produced by fiber-digesting bacteria. SCFA trigger changes in nervous system functioning and behavior. One way they do this is by activating a hormone – GLP-1 – that affects whether we feel full or hungry. In addition to SCFA, many other links between the gut and the brain have been recently discovered. Activity of the vagus nerve, a “superhighway” of information between gut and brain, is an avenue by which bacterial products in the gut affect the brain and behavior. Vagus nerve activity also affects hunger, satiety, and and body weight regulation.
SCFA are an energy source for the host, accounting for approximately 10% of dietary calories. SCFA derived from the microbial fermentation of plant fiber in the diet have a variety of beneficial effects on the host. These include improved muscle function, decreased inflammation, and decreased insulin resistance. However, when it comes to appetite, SCFA decrease appetite and induce satiety, much like ClpB. That is where the similarity ends, though. I suspect that ClpB is satiety inducing because it signals a change towards conflict in the microbial-host relationship. The relationship between fiber, SCFA, and the host is a more cooperative relationship, which I have explored in previous work with Helen Wasielewski and Athena Aktipis. The short answer to why SCFA inhibit appetite is that they contribute to dietary energy, and they reduce the need to consume extra calories by eating more. This was highlighted in a recent study has shown that mice that are germ free eat more food compared to mice colonized with gut microbes. SCFA were identified as the likely cause of this effect. In this study, germ free mice and mice with gut microbes obtained the exact same daily amount of calories overall, whether it was from eating a larger amount of chow (germ free mice) or obtaining the equivalent calories from a smaller amount of chow plus the SCFA calories (mice with microbiotas). In other words, it may be appropriate for animals that undergo fermentation of plant fiber into SCFA to consume less food overall.
We proposed in the 2014 Bioessays paper that dysphoria might be a mechanism by which microbes influence host eating behavior. It is well known that exposure to stress causes increased preference for sweet, calorie dense foods. Because energy dense diets can exacerbate anxiety, a potential vicious cycle of altered gut microbiota, altered mood, and changed dietary preference may emerge. Is there any additional evidence that microbes contribute to stress eating? This topic was recently explored by Madison and Kiecolt-Glaser who provide a mechanistic rationale for observations of altered eating behavior after stress and depression. The extent to which this mechanism is important in humans, and whether it is amenable to treatments targeting the microbiome, still awaits further testing.
In summary, the contribution of the gut microbiota to the behavior of their host, including eating behavior remains an extremely active area of investigation. Two major mechanisms of microbial influence on eating behavior – ClpB and SCFA – illustrate areas of potential conflict and cooperation, respectively, between hosts and their microbiomes. From my perspective, in both cases the host seems to be firmly in control. This may be because humans and other animals have evolved to expect manipulation from parasites, and have evolved robust defenses against interference. No defense is perfect, however, and I predict that future work will reveal additional examples of microbial manipulation of hosts involving growth-limiting nutrients and the behaviors that control them.
Categories: Uncategorized
Joe Alcock
Emergency Physician, Educator, Researcher, interested in the microbiome, evolution, and medicine

Leave a comment