This is the final installment on alternative metabolism of glucose during insulin resistance. In this post, I will describe how the addition of fucose to proteins – fucosylation – influences the risk of infection and modulates the microbiota.
Fucose is a 6 carbon monosaccharide – like glucose, mannose, fructose, and galactose – with the following chemical structure (from Wikipedia):
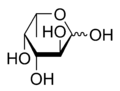
Fucosylation, which is the addition of fucose to glycans and glycoproteins, occurs by the rate limiting enzymatic activity of fucosyltransferases, such as fucosyltransferase 2 (FUT2).
In mammals, FUT2 adds fucose to N-glycans. (N-glycans have a sugar attached to a nitrogen atom, usually on the amino acid asparagine; O-glycans by contrast, link a sugar to an oxygen atom on amino acids).
The donor of fucose that is transferred by FUT2, GDP-L-fucose, requires NADPH reducing substrates that are generated by the pentose phosphate pathway. In fact, the two precursor substrates used by FUT2: GDP-L-fucose and N-glycans, both require activity of alternate metabolic pathways – the pentose phosphate pathway and the hexosamine shunt, respectively – which show increased flux during insulin resistance.
So why do mammals bother with fucose N-glycans in the gut?
Fucose N-glycation of intestinal epithelial cells affords the host protection against aberrant gut microbiota, and resulting death by sepsis. Fucosylated glycans in breast milk also gives babies increased resistance to colonization/invasion by certain gut pathogens, such as Campylobacter.
If it is so great, why do some humans lack FUT2 activity?
Interestingly, a substantial minority (20-30%) of humans carry nonsense mutations of FUT2. These polymorphisms prevent the secretion of fucose N-glycans in the gastrointestinal tract. So-called “non-secretors” without intestinal N-glycan have an increased incidence of several diseases, including type I diabetes, as described by Yang and colleagues in a 2011 Diabetes article.
Non-function of FUT2 has been shown to alter the gut microbiota and result in increased risk for auto-immune diseases such as Crohn disease, as demonstrated by Rausch et al. PNAS 2011.
The reason why selection may have favored loss of function of FUT2 was given in a recent PLOS One article. Carlsson and colleagues showed that FUT2 non-function provided protection against the Norovirus that is responsible for often fatal outbreaks of diarrheal infection. Non-functional FUT2 may also confer similar protection against Helicobacter pylori infection.
These results indicate that fucose N-glycans help prevent many bacterial intestinal infections, but increase the risk of other infections. In fact, Fumagalli and colleagues have proposed that balancing selection for resistance against various pathogens drives polymorphisms for FUT2 (as well as variation in blood type). So maybe the prevalence of FUT2 polymorphisms in human populations can be explained by infectious disease epidemiology in certain geographic regions, or bottlenecks resulting from epidemic disease…
Accumulating evidence for involvement of N-linked fucosylation in insulin resistance and diabetes:
Over 30 years ago, McMillan reported that fucose glycans are increased in the plasma of patients with diabetes. More recently Itoh and colleagues also showed an increase in fucosylated N-glycans in type 2 diabetics. Fucosyltransferases also show increased activity in human hepatoma cells exposed to elevated glucose concentrations.
In rats, insulin action helps convert the cell surface of intestinal epithelial cells from predominantly sialic acid pattern of glycation to fucose N-glycans. This change accompanies weaning and the transition to a carbohydrate rich diet. Thus insulin is important in stimulating changed intestinal glycosylation in response to diet-induced changes in gut microbiota at the time of weaning.
To recap, there is evidence that loss of function of the gene for FUT2 increases the risk of type 1 diabetes. Although FUT2 gene status is not definitively associated with type 2 diabetes, fucose N-glycans are increased among patients with type 2 diabetes .
These lines of evidence suggest that FUT2 function does not predispose to type 2 diabetes, but fucosyltransferase activity – resulting in increased production of fucose N-glycans – might be enhanced during insulin resistance.
Lipopolysaccharide – a link between insulin resistance and N-fucosylation?
Emerging data support a novel function of N-glycosylation, providing a scientifically plausible link between insulin resistance and FUT2 activity. Bacterial antigens, e.g. LPS and Vibrio cholera toxin, were been shown to induce N-fucosylation by induction of FUT2 in the mouse (Terahara et al. 2011.) In response to cholera and other environmental stresses, fucose N-gycans can be seen decorating the surface of intestinal epithelial cells:
 (fucose N-glycosylation of intestinal epithelial cells in response to Cholera toxin, Terahara et al. 2011)
(fucose N-glycosylation of intestinal epithelial cells in response to Cholera toxin, Terahara et al. 2011)
The fucose on the surface of epithelial cells feeds symbiont bacteria!
Fucose is cleaved from host glycans by enzymes expressed in commensal bacteria, e.g. Bifidobacterium and Bacteroides thetaiotaomicron. Many gram-negative pathogens, e.g. Escherichia coli, do not manufacture fucosidase, and do not regularly colonize the intestinal epithelium. As a result, fucose N-glycan production by the host results in preferential provision of nutrients to beneficial bacteria.
Lipopolysaccharide (LPS), which occurs during bacterial translocation from the gut, is a known trigger for insulin resistance. Thus, LPS can be an agonist for both insulin resistance and N-fucosylation, which has the effect of diverting a nutrient source to commensal bacteria. The diversion of some carbohydrate metabolism from glycolysis towards N-fucosylation benefits the host in this instance by feeding beneficial barrier bacteria. Selection may have favored this mutualism because commensal bacteria help prevent invasion by gut pathogens.
Just because bacteria snip off carbohydrate from N-glycans in the gut does not necessarily mean that the host generates these sugars for that reason. However, a study performed in Jeffrey Gordon’s laboratory is instructive (Bry et al. 1996). From the abstract:
“The maintenance and significance of the complex populations of microbes present in the mammalian intestine are poorly understood. Comparison of conventionally housed and germ-free NMRI mice revealed that production of fucosylated glycoconjugates and an α1,2-fucosyltransferase messenger RNA in the small-intestinal epithelium requires the normal microflora. Colonization of germ-free mice with Bacteroides thetaiotaomicron, a component of this flora, restored the fucosylation program, whereas an isogenic strain carrying a transposon insertion that disrupts its ability to use L-fucose as a carbon source did not.”
In other words, the commensal bacteria induce the production of the carbohydrate moieties that allow them to feed while the host picks up the tab. Again, Bacteroides thetaiotamicron is equipped to cleave fucose from these glycoproteins, unlike some other bacterial inhabitants of the gut. Remarkably, Bacteriodes that were unable to utilize fucose did not induce the host fucosylation of epithelial cells, suggesting a tightly coordinated signaling program between the two organisms.
Summary
N-fucosylation in the intestinal epithelium has an important function in providing protection against attachment, colonization, and invasion of certain gut pathogens. In some mammals, fucosylation accomplishes this function by preferentially feeding epithelium-associated commensal bacteria.
Some specialized viral and bacterial pathogens have evolved a capacity to use fucosylated proteins as a means of establishing infection. This counter selection has resulted in the loss of function variant of the gene encoding FUT2, and not coincidentally, is responsible for variation in human blood type. A drawback of this loss of function is an increased risk of auto-immune diseases, such as celiac disease, Crohn disease, and type 1 diabetes, each of which have been associated with altered gut microbiota. The gut microbiota, thus, is the mediator of this complex web of tradeoffs involving carbohydrate metabolism, infection risk and immune function.
Overall, these changes in glycation of host proteins are likely to play a key role in reinforcing the intestinal barrier. Future tests will be needed to confirm whether insulin resistance – and its associated shifts in carbohydrate metabolism – represent a coordinated adaptation that protects against invasive gut pathogens.
Categories: Uncategorized
Joe Alcock
Emergency Physician, Educator, Researcher, interested in the microbiome, evolution, and medicine

Since the risk of auto-immune diseases vs infection are higher in modern times, is there any way to counter the loss of fucose for homozygous non secretors? Additional, fiber to increase commensal bacteria? I understand Akermansia is a fucose degrader and 4x higher in cancerous colons.